Nanoscale energy transport
We investigate energy transport in nanoscale materials used for efficient energy transfer and conversion, and for controlling - reducing or enhancing - thermal transport in modern electronic materials and devices. At micro- and nanoscale, energy transport and conversion are determined by the dynamics of interactions among basic energy carriers such as electrons, phonons, and photons, which occurs at a time scale of femtosecond (fs, 10^-15 s) to picosecond (ps, 10^-12 s). We develop advanced femtosecond (fs) laser based high temporal resolution (~ 10 fs) experimental techniques, Raman spectroscopy-based thermometry, and high spatial resolution (~ 10 nm) scanning probe-based thermometry for the studies of nanoscale energy transfer and conversion processes.
Current research projects include (for details, see Research Projects):
- Energy transport in 2D materials and nanoelectronic devices (very thin materials of a few atomic layers bounded by van der Waals force which are used for new-generation electronic devices)
- Thermoelectric phenomena in 2D materials, which allows for in-situ cooling of electronic devices
- Polariton-enabled thermal transport as a new channel for heat dissipation and thermal management
 |
 |
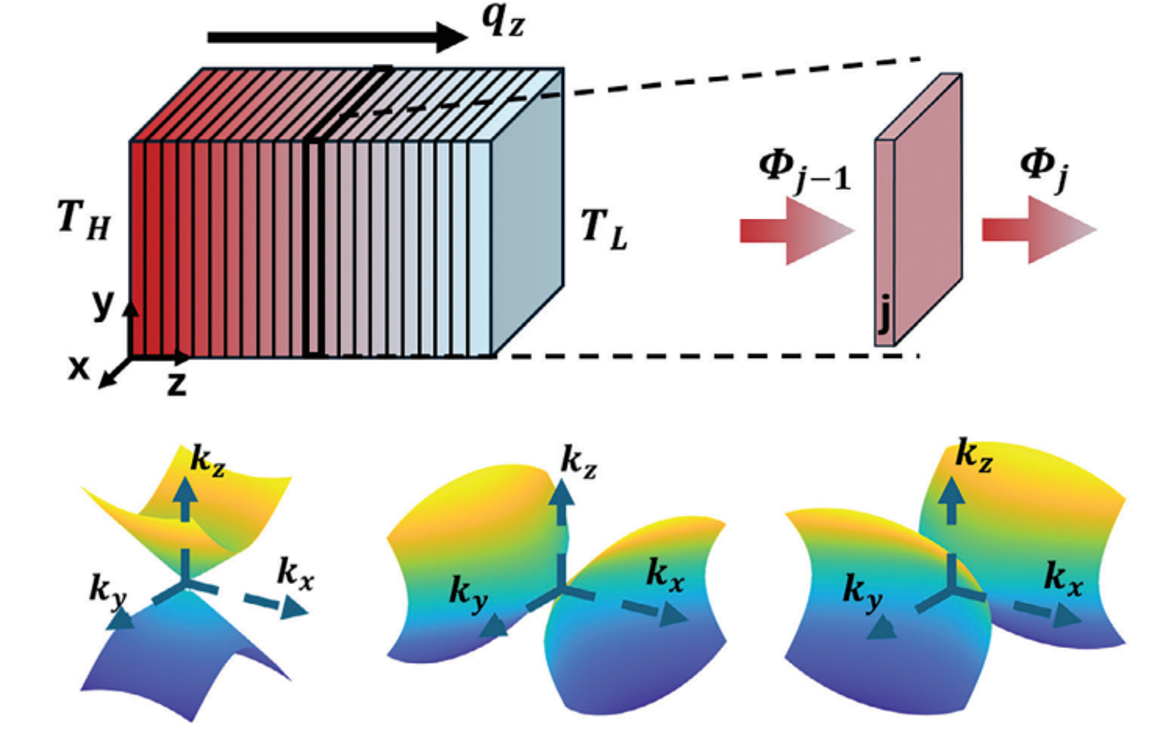 |
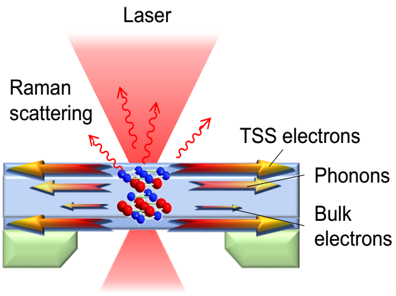
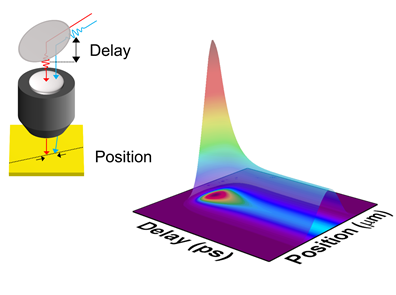
| Thermal transport in the topological insulated surface state (TSS) in topological insulator (TI) thin films. The measured in-plane thermal conductivity indicates a large thermal conductivity and a large Lorenz number L of TSS. | Spatiotemporal response of a material after laser excitation. The high resolution of the pump-probe system allows for the direct measurement of the electrical thermal transport properties of a material. | The phonon polaritons are supported by the open dispersion in hyperbolic materials. These new energy carriers creat new energy transfer channel to enhance the total heat transfer inside materials. |
Laser-based nano-optical engineering
We work on a broad range of topics related to nanoscale optical engineering. One of our major efforts is rapid 3D micro- and nanoscale printing. The main issues in 3D printing are its slow speed and accuracy. We are developing various technologies for rapid, continuous 3D nanoscale printing. We also conduct research using various machine learning tools to improve the accuracy of 3D micro- and nanoscale printing
Current projects in this area include (for details see Research Projects):
- Developing micro- and nanoscale rapid, continuous 3D projection printing
- Applications of 3D micro- and nanoscale printin
- Using machine learning tools to improve the accuracy in 3D micro- and nanoscale printing

Ultrafast laser projection nanoscale printing for fabrication of arbitrary, complex 3D microstructures in a continuous manner at high volumetric print rates.