The polymerization of polyolefins is one of the key technologies in the modern chemical industry, where a variety of catalysts are used to accelerate the rate of polymerization and control the resulting stereochemistry. Of particular interest is the use of single site catalysts which involve a metallic center that is bound to well-defined organic ligands that mediate the catalytic activity of the metal. The ultimate research objective is to understand how the structure of the catalyst effects polymerization process.
A key feature of our group’s approach has been the development of population balance models that track every species in polymerization reaction. Since polymers have large molecular weights, the number of species can be 10,000 or more, where each species can undergo a variety of fundamental reactions including initiation, propagation, different types of chain transfer, mis-insertion, recovery from mis-insertion degradation, etc. In order to accurately and rapidly produce the defining differential equation, we have developed special purpose chemical compilers where (i) the research can postulate a set of reactions, (ii) the complier automatically generates the full set of 10,000 or more ordinary differential equations (ODEs) to describe the kinetics, (iii) the ODEs are optimized for numerical solution, (iv) a set of experimental data is chosen, e.g. polymerizations at several different initial monomer concentration, and the set associated ODEs generated and (v) the best set of individual rate constants for that postulated polymerization mechanism are determined. Using this computational tools our group can access the viability of a postulated polymerization mechanism in less than a day, enabling the research to think about chemistry vs. developing a numerical solution for a particular kinetic model.
The second key feature of our group’s approach is to use rich multi-response data that includes monomer consumption via 1H NMR, evolution of the molecular weight distribution, the type of chain transfer by 2H NMR and recently the evolution of co-polymer composition via 13C NMR. We employ batch polymerization because (i) it is easy to implement experimentally and (ii) the monomer composition is changing throughout the polymerization thus challenging the model with a wide range of conditions. A viable kinetic model should be able to describe all of the multi-response data for a various initial conditions using a single set of rate constants. It might appear that with so many ODEs it would be easy to describe the experimental data; however, the opposite is true – because the equations have a very precise form due to mass action kinetics, it is very difficult to determine a viable mechanism to describe a well-defined set of multi-response data.
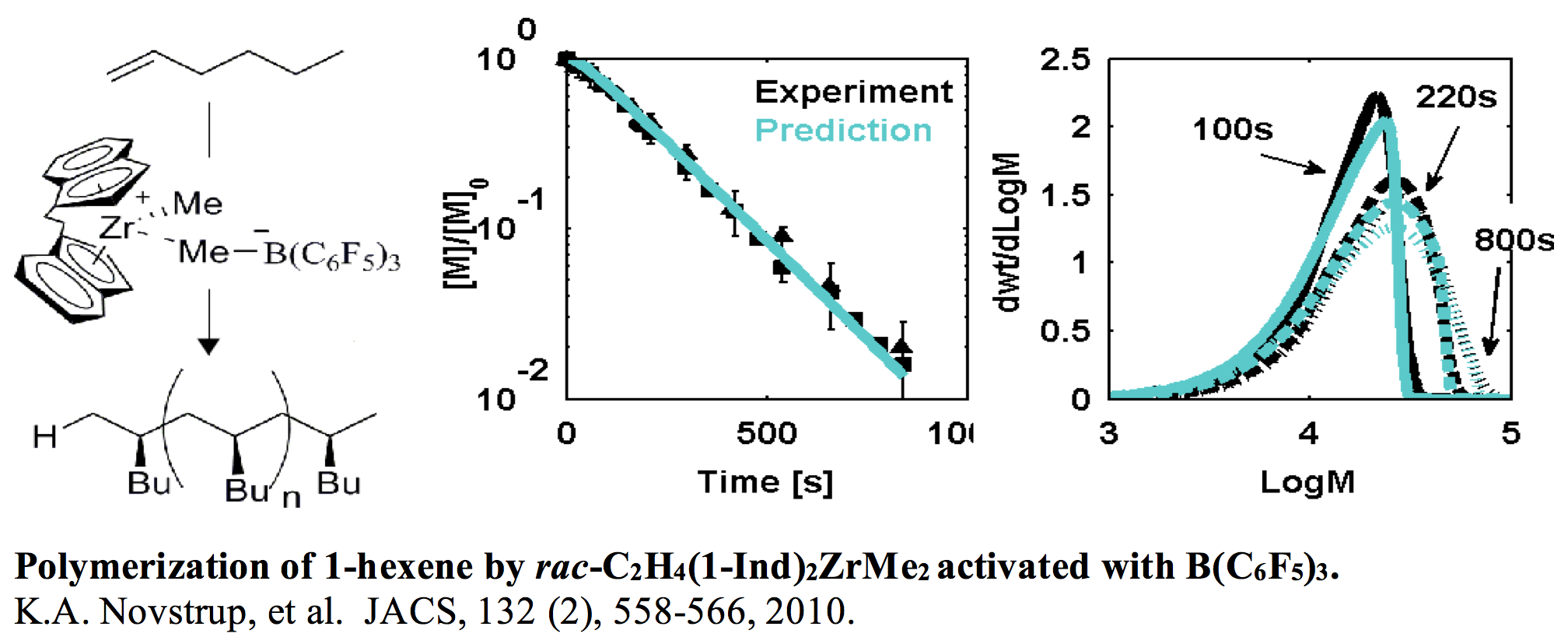
As one example consider the polymerization of 1-hexene using a bis-indenyl zirconium catalyst, where the monomer consumption and evolution of the molecular weight distribution (MWD) is shown below. Using the tools described above the catalytic mechanism was determined, e.g. only 57% of the Zirconium catalyst is active. Of particular importance is the shape of the MWD, where fitting the full shape of the distribution provides insight into the details of the kinetic mechanism.
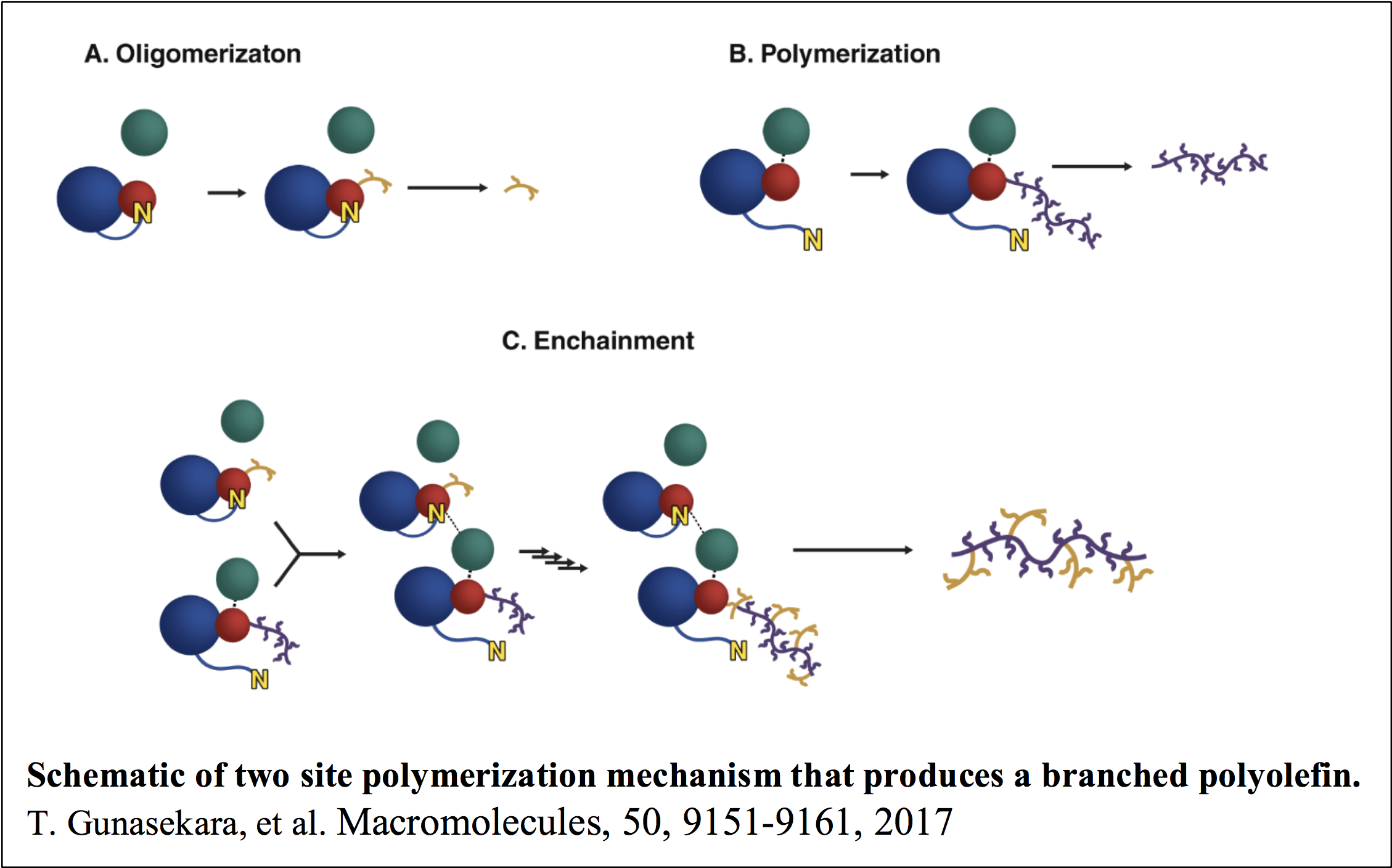
A number of single catalyst systems have been investigated for the polymerization of 1-hexene, where detailed kinetic mechanisms have been determined using the combination of multi-response data and all molecule kinetic analysis as described above. In a recent study using a zirconium amine bis(phenolate) catalyst a new type of polymerization catalysis was discovered that we call Associative Tandem Catalysis. In this system there were two catalytic sites, where the first site makes oligomers of 1-hexene and the second site polymerizes the oligomers into a branched polymer. It is interesting that the polymerization reaction is a second order reaction between the polymerization site and the oligomerization site. The identification of this type of mechanism is significant, because oligomerization is an important process in the chemical industry, where the production of polymers is undesirable because it fouls the reaction.
This work has been done in collobration with Prof. Mahdi Abu-Omar initially at Purdue and now at Univ. California – Santa Barbara Iink and Prof. Nick Delgass at Purdue.
Selected References:
1. J.M. Switzer, N.E. Travia, D.K. Steelman, G.A. Medvedev, K.T. Thomson, W.N. Delgass, M.M. Abu-Omar and J.M. Caruthers, “Kinetic Modeling of 1-Hexene Polymerization Catalyzed by ZR(tBu-ONNMe2O)Bn2/B(C6F5)3,” Macromolecules, 45, 4978-4988, 2012
2. D.K. Steelman, S. Xiong, P.D. Pletcher, J.M. Switzer, G.A. Medvedev, W.N. Delgass, J.M. Caruthers and M. Abu-Omar, “Comparison of Selected Zirconium and Hafnium Amin Bis(pehnolate) Catalysts for 1-Hexene Polymerization,” Organometallics, 32, 4862-4867, 2013
3. D.K. Steelman, S. Xiong, G.A. Medvedev, W.N. Delgass, M.M. Abu-Omar and J.M. Caruthers, “Effects of Electronic Perturbations on 1-Hexene Polymerization Catalyzed by Zirconium Amine Bis-Phenolate Complexes,” ACS-Catalysis, 4, 2186-2190, 2014.
4. P.D. Pletcher, J.M. Switzer, D.K. Steelman, G.A. Medvedev, W.N. Delgass, J.M. Caruthers and M.M. Abu-Omar, “Quantitative Comparative Kinetics of 1-Hexene Polymerization across Group IV Bis-Phenolate Catalysts,” ACS Catalysis, 6(8), 5138-5145, 2016.
5. T. Gunasekara, J. Kim, A. Preston, D.K. Steelman, G.A. Medvedev, W.N. Delgass, M.M. Abu-Omar and J.M. Caruthers, “Interaction between Two Active Sites of the Catalyst for Macromonomer Enchained Olefin Polymerization,” Macromolecules, 50, 9151-9161, 2017.